
Understanding Nutrition and Metabolic Disease
By MDA Staff | Wednesday, May 17, 2023
Mark Tarnopolsky, MD, PhD, FRCP(C), professor of Pediatrics at McMaster Children’s Hospital in Hamilton, Ontario, remembers clearly one of the first patients he saw in practice almost 25 years ago who had experienced multiple bouts of rhabdomyolysis as a child and teenager in South Africa. The patients came to Canada with a diagnosis of “PFK deficiency”, a glycogen storage disease (GSD). Dr. Tarnopolsky questioned the diagnosis based on the history of fasting and/or participation in longer-duration activities triggering the rhabdomyolysis that suggested a fatty acid oxidation disorder (FAOD) and not a GSD. Indeed, when the patient fasted for Yom Kippur and had to walk a long distance with his family to the synagogue, he would invariably get fatigued. In his early 50s he finally got the correct genetic diagnosis of carnitine palmitoyl transferase 2 (CPT2) deficiency due to a common homozygous mutation in the gene.
“We provided him with our typical FAOD advice including; no exercise when fasting or with a superimposed illness, a high carbohydrate and protein diet, a pre-exercise high carbohydrate snack (i.e., juice or fruit), and readily digestible carbohydrates every 20 minutes with sustained physical activity (i.e, sports drink, 50% diluted fruit juice, or sports gels), and identification and treatment of common deficiencies including iron, vitamin D, and vitamin B12. He has now gone 26 years without any further bouts of rhabdomyolysis given his better understanding of the disease and its treatment. In fact, he was able to cycle up to 85 kilometers with his friends on weekends in his 60s.”
Alternative fuels
Carnitine palmityl transferase deficiency is one of 11 metabolic diseases of muscle covered by MDA, in which the body’s ability to break down carbohydrates or fats for fuel is impaired. The main symptom of most of the metabolic muscle diseases is difficulty with exercise.
Nutritional strategies for regulating these diseases are based on substituting fuel sources people can use for those that they can’t. Although this sounds simple enough, in practice it can be complicated.
Carbs: First choice for energy but timing matters
Carbohydrates, fats, and protein are the three basic sources of energy. Normally, muscles are very efficient at getting the fuel they need from these sources, utilizing carbohydrates first, and then moving to fat and then to protein, if necessary.
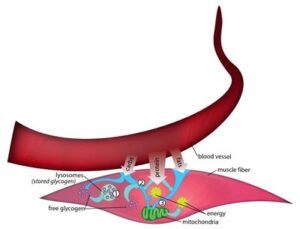
Carbohydrates include both simple sugars and complex/starches. Sugar, in the form of glucose, is transported by the bloodstream to muscle cells, enters muscle cells, and is trapped inside by the addition of a phosphate group (a phosphorus atom attached to four oxygen atoms).
When a person is exercising, sugar that comes into the cell this way is immediately broken down for energy through glycolysis. But if the person is resting, the sugar is likely mainly combined with other glucose molecules and stored in the muscle cell as a form of starch called glycogen. Enzymes are a type of protein that allows chemical reactions to take place, and all of the metabolic pathways are regulated by their activity. At rest, the glucose is synthesized into glycogen by the glycogen synthase enzyme, and at the onset of exercise, the glycogen is broken back down to glucose (glycogenolysis) by the phosphorylase enzyme and eventually used for energy through a series of enzymatically catalyzed steps in glycolysis (glucose splitting).
“When you start exercising, glycogen is the key fuel. It’s really critical for going from rest to maximal exertion,” says Ron Haller, MD, of the Institute for Exercise and Environmental Medicine at the University of Texas Southwestern Medical Center in Dallas. Dr. Haller has been awarded seven MDA grants for research projects on muscle metabolism since 1989.
Normally, when people attempt very rapid or hard exercise, glycogen metabolism in the main part of the cell kicks in right away, immediately yielding some energy itself and, at the same time, supplying pyruvate, the final product of glycogen breakdown, for a more constant, “maintenance” type of energy supply.
This maintenance kind of energy, also known as oxidative or aerobic metabolism, takes place inside mitochondria, which are bacteria-like structures (organelles) inside cells.
After glycogen molecules have been broken down and then modified to become pyruvate molecules, they can enter the mitochondria.
There, the aerobic pathway can produce about 12 times more energy per molecule of glucose (or glucose unit of glycogen) than just breaking down glycogen can. But it can’t do it as quickly.
Fat: An available but slow and steady energy supplier
Although carbohydrates are the body’s preferred source of energy, fats and, if necessary, protein (amino acids) also can be used.
“When you eat fat, like the fat around a steak, you’re eating triglycerides/triacylglycerol (TGs),” Tarnopolsky says, noting that a TG is three fatty acid (FFA) molecules bound to a glycerol molecule. After eating a meal containing TGs, the FFAs are taken up into the blood as tiny lipid balls called chylomicrons, and these can deliver the FFAs to both muscle and fat cells as energy storage. During fasting and/or longer-duration exercise, the TGs from muscle and adipose tissue can be broken down by an enzyme called hormone-sensitive lipase to yield FFAs. The adipose tissue breakdown can travel to the muscle and be taken up by fatty acids transporters and combine with the FFAs liberated from the intra-muscular TG stores to provide energy after entering the mitochondria for beta-oxidation metabolism. In order to enter the mitochondria the long-chain FFAs (the main ones used for energy during fasting and exercise) must cross the outer and inner membrane of the mitochondria through the carnitine system that involves 3 enzymes (CPT1, CPT2 and CACT). Once inside the mitochondria, the FFAs are broken down through a four-enzyme step called beta-oxidation to yield fuel for the mitochondria (the final step in oxidative metabolism).
“Although the total amount of available FFAs is much greater than stored glycogen, the speed with which energy can be liberated from FFAs is slower, and thus, one can only sustain about 50–60% of maximal exercise using FFAs vs. glycogen as an energy source.”
A boost from protein
Normally, a small amount of protein in the form of amino acids is used for metabolism, Dr. Haller says. Some of these amino acids, the building blocks of protein, can be routed to the mitochondria.
A boost from protein
“During endurance activity [such as running], you can use up to eight different types of amino acids for energy within the mitochondria, and the relative amount of amino acid oxidation increases as muscle glycogen content goes down.” Dr. Tarnopolsky says. “The maximal contribution of amino acids to energy metabolism during prolonged endurance exercise is less than 10%, and men oxidize relatively more amino acids and less glycogen than women during endurance exercise.”
Given that proteins provide important roles in the structure and function of muscle and other tissues, anything that increases protein breakdown and amino acid oxidation is akin to chopping up the furniture to heat the house. The relative amount of amino acid oxidation increases as muscle glycogen content goes down and amino acid oxidation is lower after endurance exercise training.
Putting it all together to feed the mitochondria: the “powerhouse of the cell”
During the rest-to-exercise transition, there is little oxygen available to the cell and the metabolic processes are “anaerobic.” The initial energy source comes from the adenylate kinase/myoadenylate deaminase system, phosphocreatine breakdown and anaerobic glycogenolysis>glycolysis. The adenylate kinase/myoadenylate deaminase system is a redundant and short-term buffer, and 2% of the population has zero activity of this system with no apparent issues with energy metabolism. The phosphocreatine system provides most of the energy for the first 10 seconds or so of anaerobic activity, and this is the main reason why strength and power athletes use creatine supplements. The anaerobic glycolysis system requires glycogen breakdown (glycogenolysis) and flux through the glycolysis pathway and provides most of the energy for 30–60 seconds of exercise.
Any activity longer than a minute must use the mitochondria (the powerhouse of the cell) to oxidize (metabolize with oxygen) the glucose, FFAs, and a small amount of some amino acids for energy. Mitochondria are organelles present in every cell in the human body (except for red blood cells), and they function like a furnace that combines oxygen with metabolic products to create the energy currency for the cell, ATP. The feeding of the mitochondria is similar to throwing wood on a fire. Carbohydrates and amino acids require the citric acid cycle to make “kindling sticks,” called NADH + H + and FADH2 that feed into the mitochondria at the first and second enzyme complexes of the mitochondria. NADH + H + and FADH2 are also formed by the beta-oxidation of FFAs. Ultimately, the energy from NADH + H + and FADH2 is used to make ATP in the fifth mitochondrial complex only when oxygen is present at the fourth mitochondrial complex.
Metabolic myopathies: Problems with energy derivation during exercise or other metabolic stress
The main metabolic myopathies are genetic defects in one of the enzymes responsible for the complex and elegant breakdown and subsequent use of either carbohydrates or fats. Traditionally, people had considered that defects in myoadenyate deaminase (AMPD) or acid alpha glucosidase (GAA, Pompe disease) were metabolic myopathies; however, elegant and convincing studies have clearly shown that deficiencies of either of these enzymes neither alter exercise performance nor energy metabolism in humans. As we have learned from the metabolic pathways above, these disorders typically present in the early stages of the rest-exercise transition or with bursts of more intense activity (e.g., walking up a hill). In contrast, the defects of FFA metabolism/FFA oxidation defects (FAOD) present during longer duration activity and/or with a superimposed metabolic stress such as fasting or infection. There are many genetic disorders that alter mitochondrial energy derivation pathways, and these often present with features similar to FAODs but are sometimes considered as separate entities from the metabolic myopathies as they also affect other tissues and systems.
The metabolic myopathies mainly lead to symptoms of muscle cramps or pain with activity (or metabolic stress in FAODs and mitochondrial disorders) due to the reduction in the energy currency of the cell called ATP. If the energy reduction is severe, there can be a breakdown of muscle called rhabdomyolysis, and this can damage the kidneys. The main treatment strategies for metabolic myopathies involve bypassing the metabolic block, exercise training to increase other compensatory pathways, providing alternative energy substrates, identifying and treating deficiencies that can exacerbate the disorder, and avoiding of triggering events. Unlike most of the lysosomal storage disorders where a buildup of a toxic product causes cellular damage, the only metabolic myopathy where this happens is long-chain beta-hydroxy acyl CoA dehydrogenase where the precursor to this enzyme is toxic to peripheral nerves and leads to neuropathy.
Exercise: The paradoxical best therapy
Endurance exercise training has clearly been shown to improve exercise tolerance and reduce symptoms in patients with McArdle disease. “It is well known that endurance exercise training leads to an increase in capillary blood supply to muscle and a higher capacity for fat oxidation at any given exercise intensity, and these are likely key factors explaining the benefits of exercise training in McArdle disease,” Dr. Tarnopolsky says. He cautions that patients need to start off slowly and listen to their body and only gradually increase intensity and duration as the body becomes accustomed to the training. Patients with McArdle disease should also take carbohydrate 10–15 minutes before starting activity to get into the “second wind,” which is a feeling of lower effort after a few minutes of exercise when the blood-borne fuels and oxygen are delivered to muscle. Dr. Tarnopolsky also has a number of patients with GSDs who can perform strength/resistance exercise, but he suggests that they only do 6–8 repetitions and wait 2 minutes between sets of activity. In contrast to McArdle disease and another rare form called phosphorylase b kinase deficiency, patients with glycolytic defects (i.e., GSD7, Tarui disease) should fast before and after exercise.
Patients with FAODs tolerate resistance exercise/weight training well as the FFA oxidation system is not stressed in these activities; however, Dr. Tarnopolsky advises that patients with FAODs do not do any form of exercise on days when they have a super imposed stress like a flu, cold, fever, or fasting for more than 4 hours. Patients with FAODs can perform endurance exercise but should be on a higher carbohydrate diet and take readily absorbable carbohydrate sources before and during exercise.
For further tips on exercising with a metabolic myopathy or mitochondrial disease, Dr. Tarnopolsky recommends watching videos on his team’s YouTube channel, The Neuromuscular Junction with Dr. Tarnopolsky.
Bypassing the glycogen freeway in McArdle disease
A classic example of a metabolic muscle disorder is muscle phosphorylase deficiency, also known as McArdle disease. In this condition, people lack an enzyme that is needed to break down glycogen for energy (initially anaerobic but eventually also aerobic).
 Dr. Haller says that, in his experience, people with McArdle disease have trouble doing hard exercise under any circumstances because they can’t break down glycogen, which normally yields fuel for aerobic as well as anaerobic metabolism.
Dr. Haller says that, in his experience, people with McArdle disease have trouble doing hard exercise under any circumstances because they can’t break down glycogen, which normally yields fuel for aerobic as well as anaerobic metabolism.
But they may have particular problems during the first few minutes of attempted exercise, because it takes a little time for some compensatory glucose to arrive at muscle tissue from the liver via the bloodstream.
Taking in any kind of sugar (they all get converted to glucose in the body) helps provide energy in McArdle disease because sugar can join the carbohydrate energy pathway below the point where glycogen is normally utilized.
“You’re bypassing a blocked pathway with immediate pre-exercise glucose administration,” Dr. Tarnopolsky says, comparing the glycogen metabolism pathway to a freeway that normally feeds most of its cars onto a road several miles down. “If the main freeway is shut off, you can increase a couple of feeder lanes downstream,” he explains. Starting at glucose metabolism to get around a glycogen traffic jam is taking that approach.
“If you have McArdle and can’t break down glycogen, if you take in some glucose [such as juice or, sports drink], within 15 minutes of starting exercise, it gets into the blood and allows glycolysis [glucose metabolism] to occur just as it is meant to perform in McArdle disease,” he says. However, if the glucose is taken an hour or more before exercise, then insulin will go up and the sugar is trapped as glycogen and cannot be broken down.”
Unfortunately, sugar can also be turned into fat. “The downside is, if you take in a lot of sugar without regard to the timing of exercise, four months later, you’ve gained 20 pounds. You have to match your sugar intake to your caloric expenditure and the timing of exercise,” something that usually requires close supervision by a dietician or doctor who knows a lot about nutrition.
Dr. Tarnopolsky also emphasizes that patients with defects in the glycolytic pathway (i.e, GSD 7, PGAM, etc.) should not take carbohydrates before exercise as it will worsen exercise performance since they cannot use this extra glucose, and it will inhibit the breakdown of triglycerides and proteins as alternative energy sources.
Dr. Haller recommends exercise training and a diet adequate in carbohydrates for McArdle disease. He notes that, even though McArdle patients can’t break down glycogen in muscle, they can do so in the liver, and the resulting glucose can then travel to the muscles through the circulation.
Dr. Slonim agrees that taking in sugar “in the initial phase for the first five to 10 minutes of exercise is supplying glucose at a critical period, and that’s very helpful,” he says.
He and Dr. Tarnopolsky also agree that the long-term treatment of McArdle disease should involve a high-protein diet. “When you give adequate protein, you can deal with what’s happening under these unusual conditions,” Dr. Slonim says.
Dr. Tarnopolsky has also shown that higher protein intakes lead to higher rates of amino acid oxidation that can also compensate for a reduced ability to use glycogen and generally recommends a habitual protein intake 1.5-2 times the US recommended daily allowance for myopathy patients for both metabolic and muscle maintenance reasons.
Using carbs in CPT deficiency and some beta-oxidation defects
Short- and medium-chain fatty acids can enter mitochondria on their own, but long ones can’t. They need a transport system that relies on a carrier molecule called carnitine and two enzymes called carnitine palmityl transferase (CPT) 1 and 2. Carnitine palmityl transferase deficiency and beta-oxidation defects (trifunctional protein (TFP), very long chain acyl-CoA dehydrogenase (VLCAD), and long-chain beta-hydroxy acyl CoA dehydrogenase (LCHAD) deficiency prevent people from using long-chain fatty acids.
The answer to this problem may seem as simple as giving carnitine supplements to patients with CPT2 deficiency; however, this generally only helps if the patient is carnitine deficient or in patients with a rare genetic disease called primary systemic carnitine deficiency due to SLC22A5 mutations who present in childhood with cardiomyopathy and fixed muscle weakness.
Dr. Tarnopolsky checks for total and acyl-carnitines to aid in the diagnosis of FAODs and also to identify those with low levels and only replaces those with low total carnitine levels and those rare children with SLC22A5 mutations.
Another seemingly simple and logical treatment approach for long-chain FFA disorders (CPT2, TFP, VLCAD) would be to ingest only the shorter types of FFAs. But those, Dr. Tarnopolsky notes, aren’t well tolerated by many patients.
“It’s difficult to get enough medium- or short-chain fatty acids in without getting cramps and diarrhea,” he says.
Most of them are also oily and unpalatable. Butter is a short-chain fatty acid, but eating enough of it leads to serious gastrointestinal problems.
CPT-deficient patients, are those who really benefit from a high-carbohydrate diet, Dr. Tarnopolsky says. “That’s probably where I see the most dramatic benefit. In CPT deficiency, you eat a high-carb diet to maintain glycogen stores and ingest carbs before and during exercise. Several nice studies by Dr. John Vissing’s group have shown the benefit of these strategies in patients with CPT2 deficiency, especially the high-carbohydrate diet.”
Another recent strategy is to use a supplement called triheptanoin that helps to bypass the FAODs by providing “anapleurotic” molecules to “fill up” the TCA cycle. Long-term extension studies have shown that patients with long chain FAODs had fewer metabolic crises and a shorter duration of symptoms. Dr. Tarnopolsky has found that most adults with CPT2, TFP and VLCAD deficiency do well with diet and exercise advice and they rarely have clinically significant episodes of rhabdomyolysis if they follow the advice. He reserves triheptanoin for the few who are still having symptoms in spite of adherence to diet and exercise advice.
For specific advice on nutrition in metabolic muscle disease, ask your MDA Care Center physician for a referral to a dietician or nutritionist.
Next Steps and Useful Resources
- For specific advice on nutrition in metabolic muscle disease, ask your MDA clinic physician for a referral to a dietician or nutritionist.
- Watch the Nutrition and Neuromuscular Disease MDA Engage webinar to learn more about healthy eating.
- Contact the MDA Resource Center to learn about access to dieticians and other nutritional resources. Call 833-ASK-MDA1 or email ResourceCenter@mdausa.org.
- Read The Truth About Nutrition and Neuromuscular Disease.
- Stay up-to-date on Quest content! Subscribe to Quest Magazine and Newsletter.
Disclaimer: No content on this site should ever be used as a substitute for direct medical advice from your doctor or other qualified clinician.


